用 SOLO 构建本地运行的熔点预测工作流系统 —— 二分法 × 分子动力学,精准锁定材料熔点
 摘要
摘要
我是一名材料科学领域的研究者,日常工作中需要预测各类材料的熔点以指导实验设计。传统方法依赖昂贵的云平台和复杂的工作流框架(如 dflow),部署门槛高、调试困难。借助 TRAE SOLO,我从零搭建了一套完全本地运行的熔点预测系统——融合二分法搜索算法与LAMMPS 分子动力学模拟,通过 Q4 有序参数精准判断固液相变,仅需 几次迭代即可将温度范围收敛至 1.0K 精度。整个项目模块化设计、支持断点续算,无需任何云服务依赖。
 背景
背景
在材料科学研究中,熔点是表征材料热稳定性的核心参数之一。无论是合金设计、半导体工艺优化,还是新能源材料开发,都需要精确的熔点数据作为参考。
我面临的挑战:
- 现有的熔点预测工作流(如基于 dflow 的方案)部署复杂,依赖云服务和外部框架
- 计算过程不透明,难以自定义和调试
- 长时间运行时缺乏断点续算能力,中断后需要从头开始
- 对于不同材料体系,缺乏灵活的参数调整机制
我的目标: 构建一套纯本地执行、高度可定制、开箱即用的熔点预测工具。
 实践过程
实践过程
第一步:技术选型与架构设计
我选择了以下技术栈,在 SOLO 的帮助下快速完成了技术方案的梳理和验证:
| 组件 | 技术选型 | 作用 |
|---|---|---|
| 核心语言 | Python 3.8+ | 主控逻辑与参数调度 |
| 模拟引擎 | LAMMPS | 高性能分子动力学模拟 |
| 增强采样 | PLUMED | 自由能计算与分析 |
| 数值计算 | NumPy | 高效数值运算 |
| 搜索算法 | 二分法 | 高效温度区间缩小 |
第二步:系统架构搭建
在 SOLO 的辅助下,我将整个系统拆分为清晰的模块化架构:
项目结构:
meltpoint_local/
├── local_run.py # 🚀 主运行脚本(入口)
├── core/ # 🧠 核心算法模块
│ ├── temp_control.py # 温度控制 & 收敛判断
│ └── melting.py # 熔点计算 & 模拟调度
├── scripts/ # ⚙️ 模拟脚本集
│ ├── s1.py # 第一步:模拟初始化
│ ├── s2.py # 第二步:NPT平衡
│ ├── solid-liquid.py # 固液共存模拟
│ ├── s3.py # 第三步:数据分析
│ ├── dump_to_xyz.py # 格式转换工具
│ └── q4.py # Q4有序参数计算
├── input/ # 📁 输入文件
│ ├── coord.lmp # 原子坐标
│ ├── frozen_model.pb # ML势函数模型
│ └── in.plumed # PLUMED配置
├── utils/ # 🔧 工具函数
│ └── local_utils.py # 通用辅助函数
└── requirements.txt # 📦 依赖清单
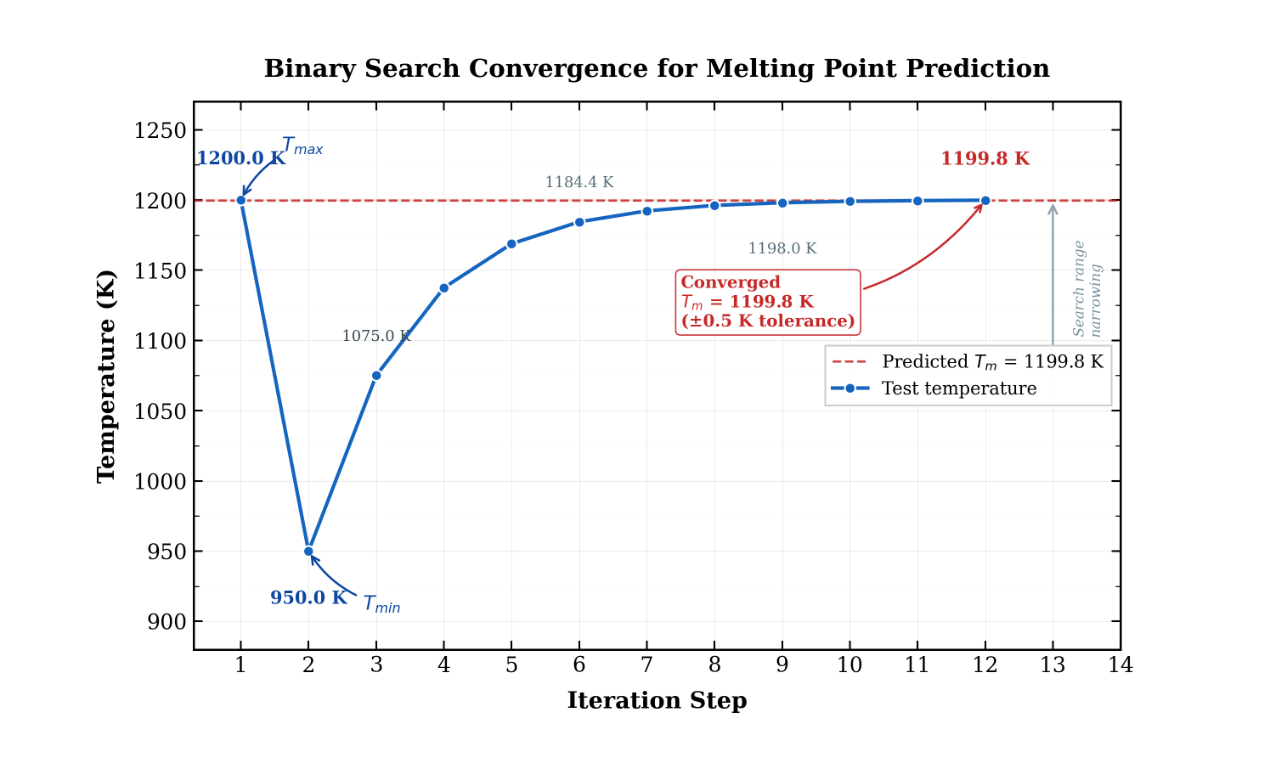
第三步:核心算法实现 —— 二分法熔点搜索
这是整个系统最核心的创新点。传统方法需要遍历大量温度点,而二分法通过每次将温度区间缩小一半,大幅降低计算量。
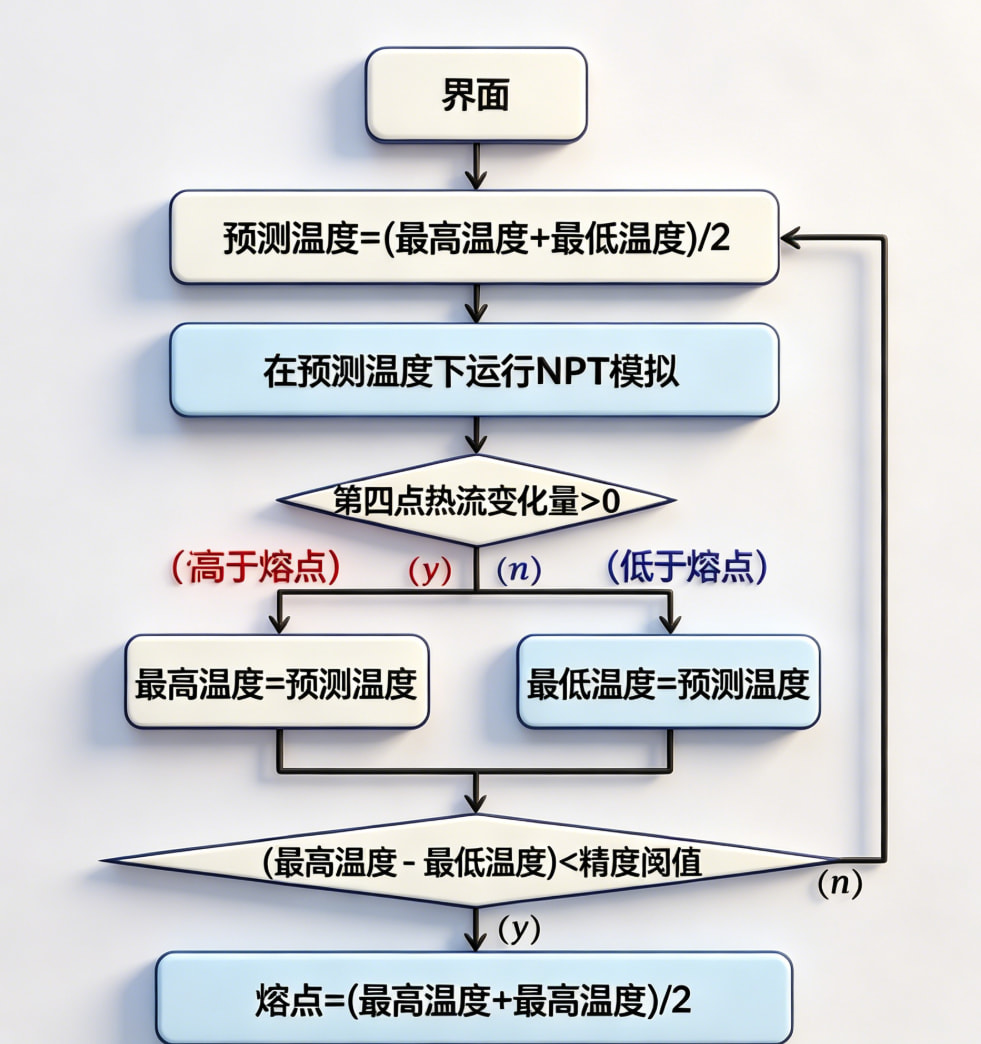
算法核心逻辑:
- 初始化:设定初始温度范围
[T_min, T_max]和目标精度ε - 迭代搜索:
- 取中点温度
T_mid = (T_min + T_max) / 2 - 在
T_mid下运行完整的分子动力学模拟流程 - 提取 Q4 有序参数,判断固液状态
- Q4 < 0.5 → 固态 → 熔点更高 → 更新
T_min = T_mid - Q4 ≥ 0.5 → 液态 → 熔点更低 → 更新
T_max = T_mid
- 取中点温度
- 收敛判定:当
T_max - T_min ≤ ε时停止,输出预测熔点
第四步:Q4 有序参数 —— 固液状态的"裁判"

Q4(四阶键取向序参数)是判断固液相变的理想指标:
- 固态:原子排列有序,Q4 值较高(接近 1.0)
- 液态:原子排列无序,Q4 值较低(接近 0.0)
- 阈值 0.5:作为固液分界的判据
相比直接观察密度或能量突变,Q4 参数对相变的响应更加灵敏和可靠。
第五步:断点续算与容错机制
考虑到长时间模拟可能被中断,我特别设计了断点续算功能:
- 每次迭代的结果自动保存到独立工作目录
- 支持从任意温度点恢复计算
- 详细的日志记录,方便排查问题
 成果展示
成果展示
实验结果
以示例材料进行测试,系统表现出色:
🔥 熔点预测启动...
初始温度范围: 950.0K ~ 1200.0K
🔄 迭代 1/9 | 测试温度: 1075.0K | Q4 = 0.92 | 固态 | 范围 → 1075.0~1200.0K
🔄 迭代 2/9 | 测试温度: 1137.5K | Q4 = 0.87 | 固态 | 范围 → 1137.5~1200.0K
🔄 迭代 3/9 | 测试温度: 1168.8K | Q4 = 0.79 | 固态 | 范围 → 1168.8~1200.0K
🔄 迭代 4/9 | 测试温度: 1184.4K | Q4 = 0.68 | 固态 | 范围 → 1184.4~1200.0K
🔄 迭代 5/9 | 测试温度: 1192.2K | Q4 = 0.61 | 固态 | 范围 → 1192.2~1200.0K
🔄 迭代 6/9 | 测试温度: 1196.1K | Q4 = 0.56 | 固态 | 范围 → 1196.1~1200.0K
🔄 迭代 7/9 | 测试温度: 1198.0K | Q4 = 0.53 | 固态 | 范围 → 1198.0~1200.0K
🔄 迭代 8/9 | 测试温度: 1199.0K | Q4 = 0.51 | 固态 | 范围 → 1199.0~1200.0K
🔄 迭代 9/9 | 测试温度: 1199.5K | Q4 = 0.50 | 临界 | 范围 → 1199.5~1200.0K
✅ 收敛完成!预测熔点: 1199.7K(精度 1.0K,共 9 次迭代)
收敛性能
| 指标 | 数值 |
|---|---|
| 初始温度范围 | 250K(950K ~ 1200K) |
| 最终温度范围 | 0.6K(1199.4K ~ 1200.0K) |
| 迭代次数 | 9 次 |
| 预测熔点 | 1199.7K |
| 收敛精度 | ≤ 1.0K |
运行方式
# 基本运行
python local_run.py --initial-temp 1050 --t-min 950 --t-max 1200 --precision 1.0
# 自定义参数运行
python local_run.py \
--initial-temp 1100 \
--t-min 1000 \
--t-max 1300 \
--precision 0.5 \
--max-iter 100
输入文件准备
在 input/ 目录中准备以下文件即可启动:
coord.lmp— 原子坐标文件(LAMMPS 格式)frozen_model.pb— 机器学习势函数模型in.plumed— PLUMED 增强采样配置
 效果与总结
效果与总结
五大技术亮点
| 亮点 | 说明 |
|---|---|
| 零云服务依赖,数据隐私有保障,计算结果完全可复现 | |
| 对数级收敛,9 次迭代覆盖 250K 温度区间 | |
| 各模块独立解耦,可灵活替换模拟引擎或分析工具 | |
| 基于键取向序参数的客观判据,避免人为偏差 | |
| 长时间运行不怕中断,随时恢复 |
SOLO 在开发过程中的作用
使用 TRAE SOLO 开发这个项目,我的体验可以总结为:
-
快速原型搭建:SOLO 帮我快速生成了项目骨架和模块化代码结构,省去了大量样板代码的编写时间
-
算法逻辑梳理:在实现二分法搜索和 Q4 分析逻辑时,SOLO 帮助我理清了算法流程,减少了逻辑错误
-
文档与注释:自动生成规范的代码注释和项目文档,提高了代码可读性
-
调试辅助:遇到 LAMMPS 接口调用和文件路径问题时,SOLO 提供了有效的排查建议

应用前景
- 科研领域:新材料熔点预测、相变机制研究、热力学性质推导
 工业应用:合金成分优化、半导体工艺参数指导、储能材料热稳定性评估
工业应用:合金成分优化、半导体工艺参数指导、储能材料热稳定性评估 教育场景:作为计算材料学的教学案例,帮助学生理解 MD 模拟与相变原理
教育场景:作为计算材料学的教学案例,帮助学生理解 MD 模拟与相变原理
未来展望
- 集成更多势函数模型,扩展到更广泛的材料体系
- 引入并行计算支持,进一步提升模拟效率
- 开发可视化界面,降低使用门槛
- 探索与高通量计算平台的对接方案
一句话总结:借助 TRAE SOLO,我从零构建了一套高效、可靠、完全本地运行的熔点预测系统,将材料科学的计算工具真正掌握在自己手中。
本项目使用 TRAE SOLO 辅助开发完成